Michigan Researchers Uncover How a 'Frankenstein' Gene Fuels Aggressive Childhood Cancer
By Anastazia Hartman | April 8Researchers in the Parolia Lab at the University of Michigan have uncovered how a single genetic event can make Ewing sarcoma—a rare and aggressive bone and soft tissue cancer that primarily affects children and young adults—more dangerous. Their findings reveal that when a gene called STAG2 is lost, the core cancer-driving protein EWS-FLI1 is redirected toward powerful genetic “on switches” that dramatically boost tumor-promoting genes.
Ewing sarcomas arise from a genetic accident causing a fusion between two independent genes, EWSR1 and FLI1. When parts of these genes break and incorrectly glue together, they form a hybrid EWS-FLI1 gene that produces an abnormal protein found in nearly all Ewing sarcomas. “It’s a bit like a Frankenstein protein,” said Abhijit Parolia, PhD, senior author of the study and an Assistant Professor of Pathology and Urology at Michigan Medicine. “This cancer-driving protein pries open segments of DNA regions that normal cells seal away and never access, turning on genes that drive cancer.”
Those segments are repetitive stretches of DNA called GGAA repeats—essentially strings of genetic sequence made up of the same four letters repeated again and again. Normally, these regions are genetic junkyards, silent and unused in healthy cells. But EWS–FLI1 turns them into potent enhancers, or molecular “on switches” that can amplify gene activity. “The fusion EWS-FLI1 protein starts reading instructions from pages of the genome that are never meant to be opened,” said Sanjana Eyunni, PhD, first author of the study. “It’s as if the cancer is accessing secret chapters of the genome to fuel its own growth.”
The new research delves into what happens when STAG2, a gene that helps organize the genome’s three-dimensional architecture, is lost. The cohesin complex that STAG2 helps form functions like a set of molecular paper clips, holding distant sections of DNA in proper alignment so that the cell’s “instruction manual” can be read correctly. Roughly 10–20% of Ewing sarcoma patients harbor STAG2 loss, and those cases are often far more aggressive, but why this happens has remained unclear, until now.
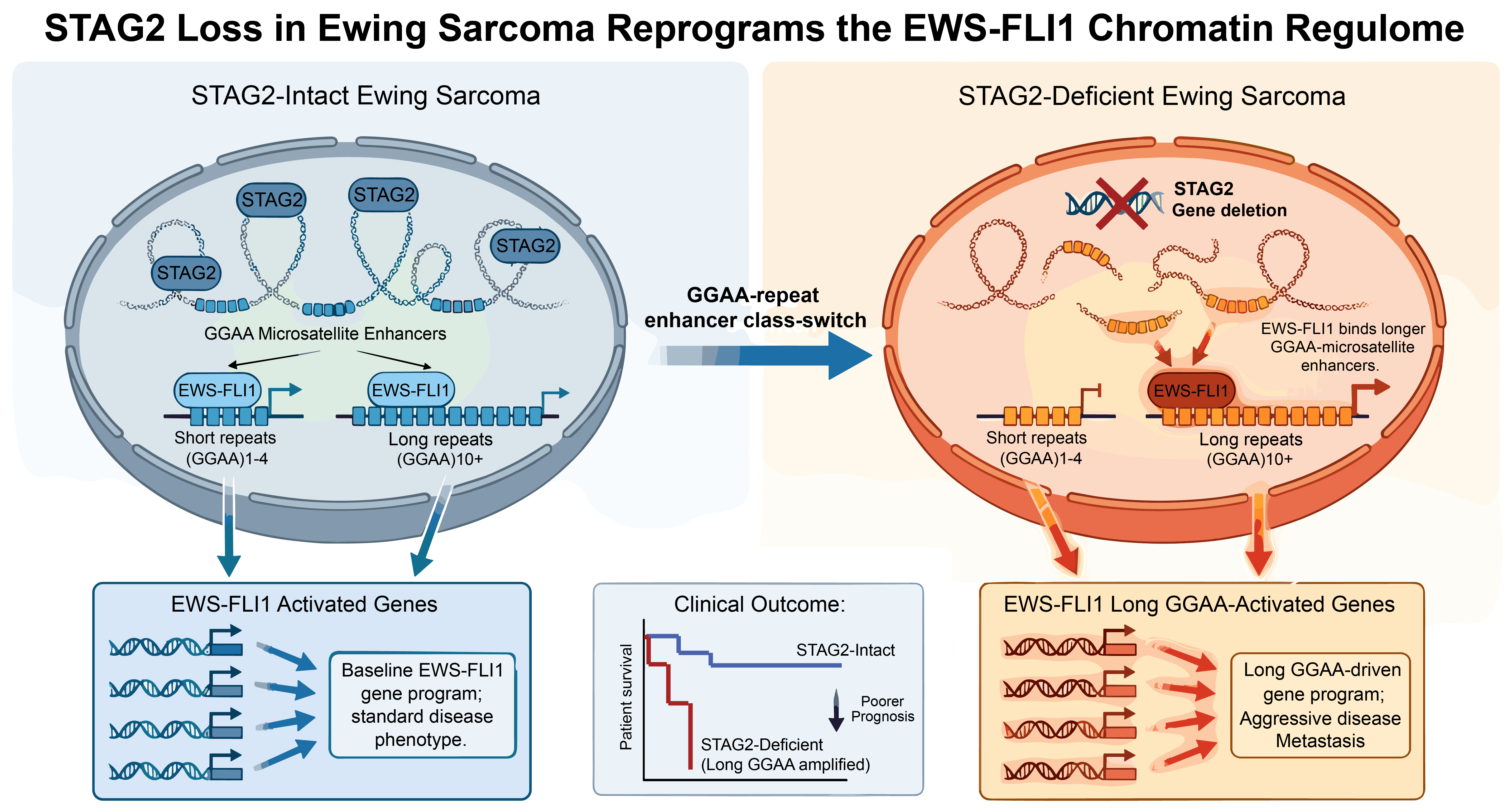
Using powerful genomic tools—including chromatin mapping and 3D genome imaging—the Michigan team discovered that loss of STAG2 doesn’t globally silence EWS-FLI1 activity. In fact, it appears as if the fusion protein doubles down on its harmful behavior. When STAG2 is gone, EWS-FLI1 abandons shorter GGAA enhancer sites and relocates to longer GGAA stretches, which act like “supercharged” switches. These long GGAA enhancers turn on a network of genes that make the cancer more invasive and difficult to treat.
The team calls this dramatic shift an “enhancer class switch,” reflecting a fundamental rewiring of gene control in STAG2-deficient Ewing sarcomas. Instead of changing the DNA sequence, the cancer changes how the genome is read—rewriting the grammar of gene regulation itself. “Tumors with this enhancer class switch show distinct gene-expression patterns that correlate with poor prognosis, pointing toward a new way to predict which patients are most at risk,” said Parolia.
Beyond its biological insights, the study offers a framework for new therapies. By targeting the architectural machinery that activates these expanded GGAA regions, researchers hope to disrupt the cancer’s abnormal DNA-reading pattern. As Parolia summarized, “Understanding how this Frankenstein protein rewires the genome in aggressive tumors gives us new leverage points to design smarter treatments and, ultimately, improve outcomes for young patients facing this devastating disease.”
__
Read the full article on the PNAS website.
DOI number 10.1073/pnas.2537425123
__
This work was funded by the National Cancer Institute Outstanding Investigator Award (R35-CA231996, A.M.C.), NIH K00 Fellowship (K00-CA245825, A.P), Rogel Fellowship (A.P.), and the V Foundation for Cancer Research–Scholar Award (V2024-020, A.P.).
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER