Study from Lieberman Lab Published in Acta Neuropathologica
By Elizabeth Walker | April 28 2020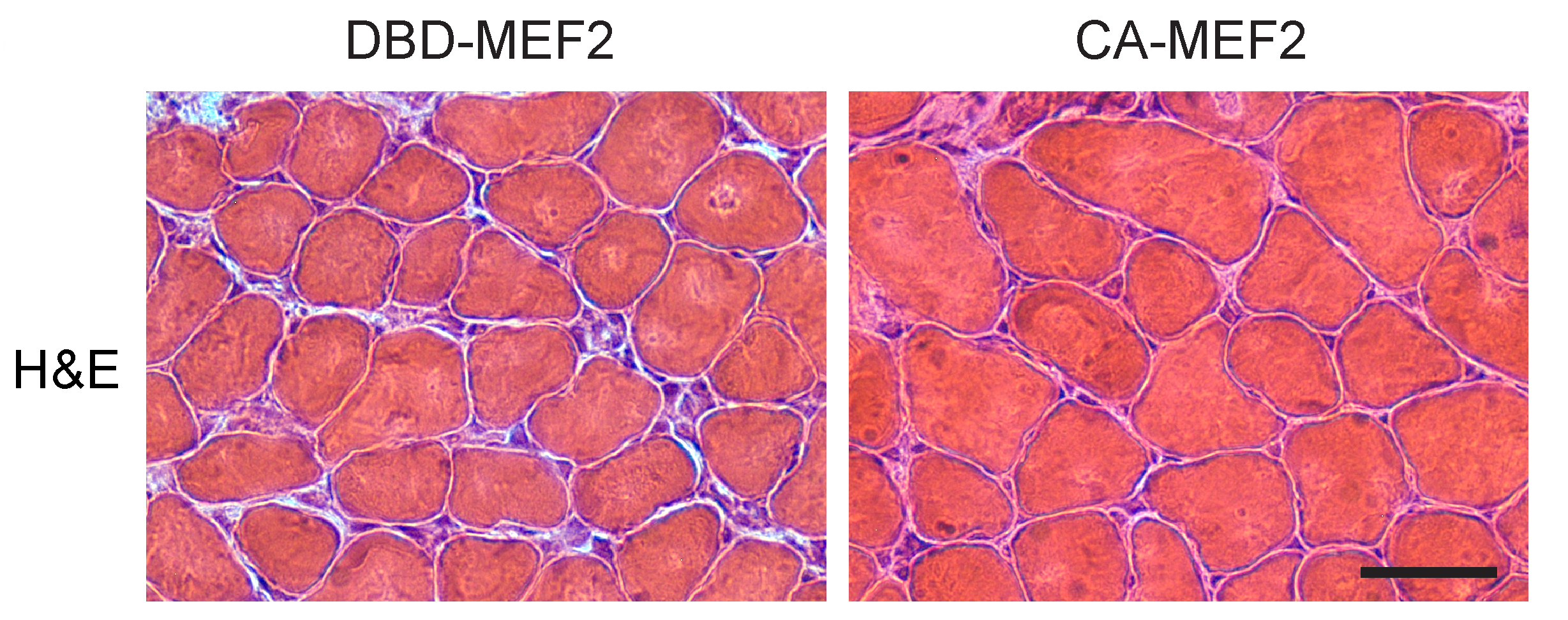 A new study from the Lieberman Laboratory’s Samir Nath, et al, has discovered that MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease.
A new study from the Lieberman Laboratory’s Samir Nath, et al, has discovered that MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease.
Kennedy’s Disease, also known as Spinal and Bulbar Muscular Atrophy (SBMA), is a disease caused by an inherited gene mutation that results in muscle and brain cell dysfunction. Patients progressively lose muscle mass until they are unable to walk without assistance. Ultimately, muscles involved in breathing can also be affected. As a result, a leading cause of death in these patients is pneumonia. To date, there are no FDA approved treatments for SBMA.
Researchers have long wondered what drives loss of muscle mass in SBMA. Previous work from the Lieberman Lab found that a cellular waste incinerator called the ubiquitin-proteasome system, a key driver of muscle loss downstream of fasting, paralysis, and cancer, isn’t increased in SBMA muscle. This surprising finding led them to attempt to answer the question: Why is muscle loss occurring in SBMA?
The paper, MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease, published in Acta Neuropathologica, details the search for possible causes of muscle wasting in SBMA, which led to the identification of a novel therapeutic target in disease.
Initially, many of the lab’s results in SBMA mice were opposite expectations in a disease of muscle wasting- illustrating that something unique is happening in SBMA muscle. To the researchers’ surprise, proteins that tag muscle for destruction were reduced rather than increased, which should increase muscle size rather than lead to muscle wasting. Further, signaling pathways that drive muscle growth were increased rather than decreased, which should again increase muscle size rather than cause muscle wasting. Finally, the stem cells that generate new muscle were intact and capable of regenerating damaged muscle.
These findings suggested that the muscle may be compensating for something by driving these pathways towards sparing muscle, but the team had yet to identify a mechanism of muscle wasting. Using bioinformatic analyses, they landed on a key regulator of muscle balance, a “transcription factor” called Myocyte Enhancer Factor 2 (MEF2). They found that mice which carry the mutation that causes SBMA lose MEF2 function over time as symptoms develop.
Through a series of studies, it was found that the mutant protein which causes SBMA, called polyglutamine androgen receptor, can directly interact with MEF2, and the authors suggest that this may be how it impairs MEF2 activity. Surprisingly, they found that another protein which drives a related disorder, called Huntington’s Disease, can also interact with MEF2 and cause a very similar loss of MEF2 targets in mouse and cell models. Importantly, skeletal muscle biopsies of SBMA patients also demonstrated signs of MEF2 impairment, suggesting this mechanism is not unique to mouse models but also occurs in people. Finally, when they restored MEF2 function in SBMA mice using a specialized technique called electroporation, it significantly rescued muscle mass. These findings are very exciting, as they suggest MEF2 activity is a novel therapeutic target in SBMA and that further research is warranted to determine its therapeutic potential.
This work raised several new questions the team is now investigating, including how long-lasting are the effects of restoring MEF2 activity? Further, because the techniques used to restore MEF2 activity aren’t currently feasible in humans, small molecules which target MEF2 and increase its activity need to be identified to translate these results to the patient population.
The lab is excited to provide new mechanistic insight behind muscle wasting in SBMA and hopes that these results will aid in the development of a therapeutic that can one day ameliorate muscle wasting in this disease. Read the entire paper at Springer Link.
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER
 ON THE COVER
ON THE COVER